Nicotinamide mononucleotide (NMN), a precursor to the vital cellular cofactor NAD+, is gaining strong human data as a potential ally against age-related decline in muscle and liver health.
NAD+ (nicotinamide adenine dinucleotide) is essential for energy production, DNA repair, and the activity of longevity-related enzymes called sirtuins. NAD+ levels naturally decline with age, and this drop has been linked to reduced mitochondrial function, increased inflammation, and metabolic dysfunction in multiple tissues.
NMN is a direct NAD+ precursor that can raise NAD+ levels when taken orally, as shown in numerous animal and early human studies. By restoring NAD+ availability, NMN may help support healthier metabolism, better energy handling, and improved cellular repair in organs such as muscle and liver.
New Multi-Study Evidence: Muscle Benefits
A recent meta-analysis from Wuhan University pooled data from 13 randomised controlled trials (9 independent studies) involving 412 middle-aged and older adults from Japan, China, and the United States. Participants took between 250 and 1250 mg of NMN daily for 4–24 weeks, allowing researchers to assess real-world dosing and duration.
In eight trials that measured gait speed, a key marker of mobility and functional aging, NMN supplementation increased walking speed by roughly 1 ft/s on average, although individual responses varied. Gains were notably larger in participants under 60, suggesting that earlier intervention may yield greater muscle-performance benefits.
Interestingly, grip strength, another common muscle measure, showed little change overall, with some improvement only in adults over 60. This pattern suggests that NMN may preferentially enhance dynamic performance (like walking) rather than raw strength, at least over short- to medium-term supplementation.
Liver Health and Metabolic Effects
The same meta-analysis examined standard markers of liver function, AST (aspartate aminotransferase) and ALT (alanine aminotransferase), across six clinical trials. While NMN did not significantly alter AST, it did significantly reduce ALT levels, particularly when taken for less than 10 weeks or in participants under 60.
Elevated ALT is often a sign of liver strain or injury, so reductions may indicate improved liver resilience or lower underlying inflammation. The analysis also found that lower doses of NMN had the strongest effect on insulin resistance (measured by HOMA-IR), hinting that modest, sustained dosing could support metabolic health without requiring high intakes.
However, not all trials were uniformly positive. A Harvard-led RCT using a higher dose (2 grams per day) in overweight and obese adults did not find improvements in liver enzymes, insulin resistance, or muscle strength measures such as stair climbing. This contrast supports the idea that dose, duration, and participant characteristics matter greatly when evaluating NMN’s benefits.
Safety, Limitations, and Practical Takeaways
Across the pooled trials, NMN was generally well tolerated, with no serious adverse events reported, supporting its reputation as a safe supplement for middle-aged and older adults. Researchers concluded that NMN shows “positive efficacy” in enhancing muscle function, reducing insulin resistance, and lowering liver aminotransferase levels, calling it an “encouraging and considerable” candidate for anti-aging treatment.
At the same time, the authors emphasised that the existing studies are relatively small, short in duration, and variable in quality, making larger, multi-center RCTs essential before firm clinical recommendations can be made. NMN should be viewed as a promising adjunct, ideally combined with exercise, a balanced diet, and good sleep, rather than a standalone solution.
For individuals considering NMN, sensible steps include:
References
Metformin, a decades-old diabetes medication, is emerging as a surprising contender in the fight against age-related vision loss. Recent research suggests it may help protect the aging retina and slow changes that eventually impair sight.
Age-related macular degeneration (AMD) and other retinal diseases are leading causes of irreversible blindness in older adults. As we age, oxidative stress, chronic inflammation, and abnormal blood vessel growth damage the delicate tissues at the back of the eye, especially the macula, which is critical for sharp central vision.
Current treatments can help in advanced stages, such as injections for “wet” AMD, but there are no widely available therapies that reliably prevent or halt early retinal aging. That gap has driven interest in repurposing safe, well-known drugs such as metformin.
Metformin: More Than a Diabetes Drug
Metformin is one of the most prescribed medications worldwide for type 2 diabetes, where it improves insulin sensitivity and lowers blood sugar. Over the last decade, it has also been studied for “anti-aging” benefits, including potential protection for the heart, brain, and eyes.
In the retina, preclinical work shows that metformin exerts anti-inflammatory, antioxidant, and antiangiogenic effects meaning it can reduce damaging inflammation, oxidative stress, and abnormal vessel growth. Animal and cell studies report that metformin helps preserve retinal structure, normalises key molecules like VEGF and inflammatory cytokines, and supports healthier retinal vasculature.
New Evidence: Metformin and Age-Related Vision Loss
A growing number of human studies are now backing up these lab findings. A recent five-year, image-based study in people with type 2 diabetes found that those taking metformin had about 37% lower odds of developing intermediate AMD compared with non-users, even after adjusting for other factors. This suggests metformin could help delay a critical stage of macular degeneration before major vision loss occurs.
Systematic reviews and meta-analyses pooling millions of participants also report that metformin use is associated with reduced odds of AMD overall. Additional observational work indicates that metformin may slow progression to both dry and wet forms of AMD and may also offer protection against other retinal diseases such as diabetic retinopathy and glaucoma.
Because metformin activates AMPK and influences cellular energy and autophagy pathways, researchers believe it may help retinal cells better manage waste products and resist stress over time.

(Liu et al., 2025) Metformin Improves Visual Cortex Information Processing. Aged mice (red) exhibit lower information processing than young mice (green), particularly with larger populations of neurons. However, aged mice treated with metformin (blue) exhibit even higher levels of information processing than young mice.
What This Means for Patients
Metformin is not yet an approved treatment for AMD or other age-related eye conditions, and experts emphasise that more randomised clinical trials are needed. Ongoing and planned studies are testing whether oral metformin can directly slow retinal degeneration in specific diseases such as ABCA4 retinopathy and geographic atrophy.
For now, the findings are encouraging but not a license for self-prescribing. Metformin can cause side effects and is typically used under medical supervision, especially in people without diabetes or with kidney issues.
If you are concerned about aging and vision:
Metformin’s track record, safety profile, and emerging retinal data make it one of the most promising drug candidates in the quest to slow age-related vision decline offering hope that future therapies may not only treat, but also delay, the onset of blinding eye diseases.
Dosage
Model: 12-month-old male C57BL/6J mice
Dosage: 250 mg/kg of metformin (corresponding to a human dose of 1200 mg/day) orally administered for three weeks
References
Modern life often feels like a constant race, deadlines, notifications, worries and our bodies pay the price in the form of chronic stress and accelerated aging.
Researchers from the University of California, San Francisco, Columbia University, and the University of Denver propose a powerful idea: chronic stress may speed up aging by stealing energy from our cells’ restorative work.
When we perceive a threat, our bodies enter a “survival mode” driven by the sympathetic nervous system (SNS), releasing hormones like cortisol and adrenaline that increase heart rate and blood pressure and trigger waves of inflammatory cytokines.
All of this is energetically expensive. Producing stress hormones and cytokines requires large amounts of ATP (adenosine triphosphate), while the faster heartbeat and heightened alertness also consume extra energy energy that might otherwise have gone toward cellular repair and maintenance.

What Energises the Heart? Listed are the molecules that fuel the heart, the most metabolically active organ in the body. What fuels the heart are the substrates mitochondria utilise, along with oxygen, to produce ATP. As depicted, the heart primarily uses fat (Fatty acids, 40-70%) to produce ATP, followed by sugar (Glucose, 20-30%).
Mitochondria: Where Stress and Aging Meet
Mitochondria are the tiny power plants in our cells that use oxygen to make ATP, and they are increasingly viewed as central players in the aging process.
If chronic stress constantly demands more ATP for threat responses, mitochondria become taxed, less efficient, and more prone to generating damaging byproducts that contribute to cellular aging.
Cellular restoration the set of processes that repair molecular damage, maintain organ function, and delay biological aging depends heavily on healthy mitochondria.
When stress diverts energy away from restoration and toward “threat management,” damage accumulates faster, potentially leading to premature aging at the cellular level.

Illustration of healthy vs dysfunctional mitochondria showing factors like physical activity, toxins, healthy food, and stress
The Autonomic Nervous System: Stress vs. Safety
The autonomic nervous system (ANS) has two main branches that constantly shape our internal state.
The SNS prepares us for action (fight-or-flight), while the parasympathetic nervous system (PNS), largely driven by the vagus nerve, supports rest, digestion, and deep repair.
Our breathing pattern is one of the most direct “signals” the brain uses to gauge whether we are safe or in danger.
Fast, shallow breaths tend to reflect or reinforce a threat state, whereas slow, deep breathing promotes parasympathetic dominance essentially telling the body, “It’s safe to relax.”
Slow Breathing: A Simple, Powerful Tool
Breathing at about six breaths per minute (for example, inhaling through the nose for a count of six and exhaling through the nose for a count of six) has been linked to improved oxygen use by mitochondria, lower blood pressure, and healthier heart rate patterns.
Contemplative practices such as meditation, prayer, and breath-focused relaxation often naturally slow breathing and are associated with better stress resilience and health outcomes.
Over time, daily practice can strengthen resting parasympathetic dominance, making us less reactive to stress and potentially less vulnerable to premature biological aging.
Training the Mind: Thoughts as Hidden Stressors
Not all threats are physical; repetitive worry and negative thinking can activate the SNS just as effectively as real danger. Ruminating on past regrets or future fears keeps the body in a chronic “threat arousal” state, draining energy and fueling stress-driven aging.
Slow, mindful breathing helps us notice these thought patterns and interrupt them before they spiral.
A large analysis of 135 studies found that better emotional regulation including managing negative feelings is linked to higher cardiac vagal control, a marker of stronger parasympathetic tone and greater nervous system balance.

By regularly practicing slow breathing, contemplative techniques, and thought awareness, we create more frequent states of deep rest in which mitochondria can channel ATP into cellular restoration.
In contrast, living in constant stress mode reduces this restorative time, increasing the risk of premature aging at the biological level.
Practical Breathing Exercise
Cromwell et al., theory on stress, mitochondrial energy allocation, and cellular aging
A new line of research suggests that one small molecule released during exercise could play a big role in keeping our brains sharp as we age.
Aging is a major driver of cognitive decline, dementia, and neurodegenerative disease, yet effective interventions remain limited.
Aerobic exercise stands out as one of the most powerful lifestyle tools we have, improving memory, mood, sleep, and slowing neurological decline in older adults and animal models.
Scientists now know that during exercise, our muscles, liver, and even neurons release signalling molecules called exerkines that help coordinate beneficial changes throughout the body.
One exerkine in particular, β-hydroxybutyrate (BHB), is emerging as a key player linking physical activity to better brain health.
Meet β‑Hydroxybutyrate: The “Exercise Ketone”
BHB is the most abundant ketone body made in the liver from fats during fasting, low-carbohydrate intake, or aerobic exercise, when blood glucose is relatively low. Both the brain and muscles can use BHB as an efficient fuel, and it has been studied for potential benefits in conditions like neurodegenerative disease, heart failure, and liver disorders because of its anti-inflammatory and neuroprotective properties.
In a new study published in the Journal of Sport and Health Science, researchers from Shanghai University of Sport showed that a single one-hour treadmill session in 19‑month‑old mice (roughly human age 60) more than doubled circulating BHB three hours after exercise, before returning to baseline within 24 hours.
These findings confirm that even in older organisms, aerobic exercise robustly raises BHB levels.

Exercise more than doubles circulating levels of β-hydroxybutyrate. Old mice that underwent an hour of exercise (blue line) exhibited more than doubled circulating β-hydroxybutyrate compared to non-exercised old mice (red line).
Exercise, BHB, and Better Memory
To test whether this surge in BHB actually mattered for the brain, the researchers put older mice on a 16‑week aerobic program—running one hour a day, five days per week.
After the training period, the animals performed better on memory tests, confirming that long‑term aerobic exercise enhances cognition in aging brains.
The team then asked a critical question: could BHB alone mimic these benefits? They injected old mice with BHB five times a week for 16 weeks at a dose of 200 mg per kg of body weight. Supplementation nearly tripled circulating BHB within 15 minutes and, remarkably, produced similar improvements in memory assessments to those seen with exercise.
In a follow‑up experiment, mice were genetically modified to lack an enzyme needed to produce BHB, and these animals failed to gain the same cognitive boost from exercise as normal mice. This suggests BHB is not just a by‑product, but a necessary mediator of at least part of exercise’s brain-protective effects.
What This Could Mean for Human Brain Aging
Human studies show that boosting ketone availability through fasting, ketogenic diets, or exogenous ketones can enhance brain network stability in younger adults, hinting at a protective effect that may also apply to aging brains.
Researchers believe BHB could help guard against age-related cognitive decline, mild cognitive impairment, and possibly Alzheimer’s disease, but clinical trials in older adults are still needed.

For now, the most proven strategy remains regular aerobic exercise, which naturally raises BHB and provides many other systemic benefits.
However, for people interested in “hedging their bets,” BHB supplements—often sold as ketone powders—are already commercially available, typically costing around $30–$45 for a month’s supply.
Anyone considering supplementation should remember that the exciting data so far come primarily from animal studies, not large human trials.
Combining consistent exercise, a nutrient-dense diet, good sleep, and, where appropriate, physician-guided use of BHB supplements may offer a promising, science-informed path toward maintaining a sharper, healthier brain with age.
References
A growing body of research suggests we may be looking at aging the wrong way by focusing on many separate “problems” instead of a central driver: our mitochondria, the tiny power plants inside our cells that generate energy.
In 2013, scientists outlined nine “hallmarks of aging,” including genomic instability, loss of proteostasis, and mitochondrial dysfunction, to describe the main biological processes that drive aging.
A decade later, three more were added chronic inflammation, disabled macroautophagy, and dysbiosis bringing the total to twelve interconnected hallmarks.
Not all researchers agree that these hallmarks are equally important. A growing camp argues that mitochondrial dysfunction may be the upstream trigger that aggravates many of the others, making it one of the most crucial factors in how and why we age.
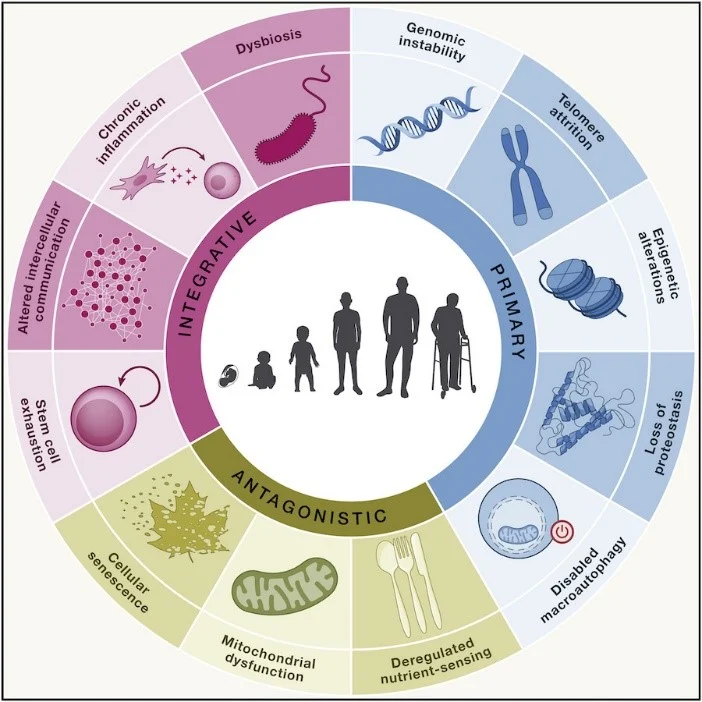
Why Mitochondria Matter So Much
Mitochondria produce ATP (adenosine triphosphate), the universal energy currency that powers nearly every cellular function.
Cells such as those in the heart can contain thousands of mitochondria, reflecting the immense energy needs of highly active tissues.
As mitochondria generate ATP, they also produce reactive oxygen species (ROS), highly reactive molecules that in small amounts act as important signaling messengers.
When mitochondria become dysfunctional, ROS production can surge, creating oxidative stress that damages DNA, proteins, and lipids, and feeds directly into several hallmarks of aging such as genomic instability, cellular senescence, and chronic inflammation.
Lifestyle: The First Line of Mitochondrial Defense
Two everyday habits overeating and being sedentary have an outsized impact on mitochondrial health.
Overeating, especially calorie-dense processed foods high in refined sugars and fats, can overwhelm mitochondria and drive excessive ROS production.
Whole foods like fruits and vegetables, which are nutrient-rich and lower in calories per volume, better support efficient metabolism and help protect mitochondria from oxidative damage.
A sedentary lifestyle reduces mitochondrial biogenesis, the creation of new mitochondria, and often leads to more body fat and less muscle mass.
With fewer mitochondria handling excess calories, oxidative stress can rise even further, accelerating age-related damage and contributing to stem cell exhaustion and tissue degeneration.
Targeted Nutrients for Mitochondrial Support
Beyond diet and movement, certain nutritional compounds may help support mitochondrial function as we age.
NAD+ (nicotinamide adenine dinucleotide) is essential for mitochondrial ATP production and fuels protective enzymes called sirtuins; its levels tend to be lower in older and obese individuals.
Precursors such as nicotinamide, nicotinamide riboside (NR), and nicotinamide mononucleotide (NMN) can raise NAD+ levels and are being explored as longevity-supporting ingredients.
Urolithin A has been shown to stimulate mitophagy, the process that selectively clears out defective mitochondria to maintain a healthy mitochondrial network.
Combining NAD+ boosters with mitophagy activators, as in multi-ingredient nutraceuticals like Restorin, may offer a synergistic strategy for targeting mitochondrial dysfunction.
Rethinking Aging: Energy First
Aging is almost certainly multifactorial, and scientists still debate whether mitochondrial dysfunction is the single “root cause” of degenerative aging.
However, because every hallmark of aging ultimately depends on adequate ATP, maintaining mitochondrial health through smart nutrition, regular physical activity, and potentially targeted supplements may be one of the most powerful ways to increase our chances of living longer, healthier lives.
Reference: https://pubmed.ncbi.nlm.nih.gov/39866226/
Reference: https://pubmed.ncbi.nlm.nih.gov/39923381/
Apigenin, a natural compound found in various fruits and vegetables, has recently been studied for its potential therapeutic effects against certain cancers, including leukemia and melanoma. Two notable studies have explored how apigenin may combat these diseases through different mechanisms.
In the first study, researchers investigated apigenin's impact on acute lymphoblastic leukemia (ALL) cells. They discovered that apigenin could induce a type of cell death known as apoptosis in these cancer cells. This process was facilitated by activating the AMP-activated protein kinase (AMPK) pathway, which plays a crucial role in cellular energy regulation. Activation of AMPK led to ferroptosis, another form of cell death characterized by iron-dependent lipid peroxidation, effectively reducing the viability of leukemia cells.
The study also highlighted that apigenin's induction of ferroptosis was associated with increased reactive oxygen species (ROS) production and depletion of glutathione, an important antioxidant in cells. By disrupting the balance of oxidative stress and antioxidant defenses, apigenin created an environment unfavorable for leukemia cell survival.
In the second study, scientists focused on apigenin's potential against melanoma, a serious form of skin cancer. They developed a specialized delivery system called apigenin-loaded invasomes to enhance the compound's ability to penetrate the skin and reach melanoma cells effectively. Invasomes are vesicles designed to improve the delivery of active substances through the skin barrier.
Laboratory tests demonstrated that these apigenin-loaded invasomes significantly inhibited the growth of melanoma cells. The treatment led to a reduction in cell proliferation and induced apoptosis in the cancer cells. This suggests that the enhanced delivery system could make apigenin a more effective option for topical treatment of melanoma.
The researchers also noted that the invasome formulation improved the stability and skin permeation of apigenin, addressing previous challenges related to its delivery and effectiveness. This advancement could pave the way for developing new topical therapies for skin cancers, utilizing natural compounds like apigenin.
While these findings are promising, it's important to recognize that both studies were conducted in controlled laboratory settings. Further research, including clinical trials, is necessary to determine the safety and efficacy of apigenin-based treatments in humans. Nonetheless, these studies contribute valuable insights into the potential use of natural compounds in cancer therapy.
In summary, apigenin shows potential as a therapeutic agent against leukemia and melanoma by inducing cancer cell death through mechanisms like apoptosis and ferroptosis. Innovative delivery systems, such as apigenin-loaded invasomes, may enhance its effectiveness, particularly for skin-related cancers.
Continued research is essential to translate these findings into practical treatments for patients.